
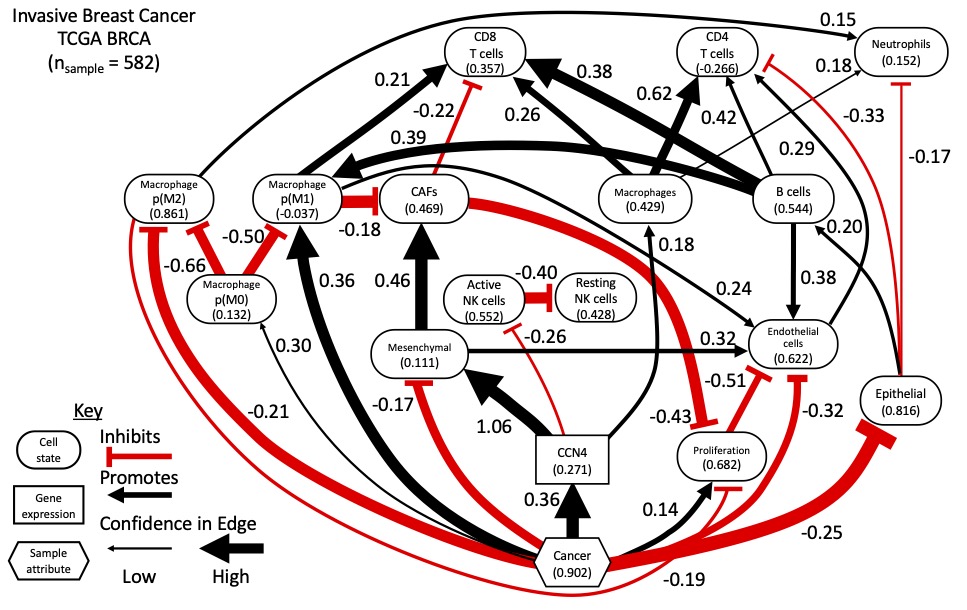
Typically scientific conferences are where one publicly professes scientific ideas. In expressing those ideas, you open those ideas to critique. With the limitation of conferences due to COVID-19 in recent years, opportunities for scientific discourse and the exchange of ideas has been limited. Also in recent years, we have been working towards developing data-driven methods for inferring how cellular networks change in disease. We recently published a paper in Nature Communications describing the approach [Klinke et al., 2022]. In particular, we have focused on identifying how Cell Communication Network Factor 4 (CCN4) that is secreted by malignant cells alters the cellular composition and functional orientation of immune and stromal cells within the mammary gland during oncogenesis. Using the prevalence of these cellular features predicted from bulk RNAseq data obtained from over 500 homogenized normal and malignant human mammary tissue samples, we used Bayesian network inference to predict the causal network shown above, assuming that the causal Markov condition applies.
One of the challenges with this approach involving human data is that the ground truth is not available. In contrast to measuring engineered systems created by the human hand or data sets generated synthetically where the relationships among components of the system are known, the generative causal relationships, that is the ground truth, in human (patho-)biology are not completely known. While we did validate aspects of the predicted causal network using syngeneic mouse tumor models, we can envision more rigorous testing involving a project with three aims. The first aim is to generate an analogous dataset using a model system of breast cancer where the ground truth can be independently measured and the predicted causal relationships can be externally validated. Here, the ground truth means measuring the cellular features using independent experimental means and generating a time course data set, which is the traditional approach to establish causal relationships. The second aim is to validate the cellular features predicted by deconvoluting the bulk transcriptomics data set. The third aim is to externally validate a subset of the causal relationships predicted from the dataset obtained in the first aim. There are a number of questions that people have raised about this approach. In the text that follows, I’m going to try to address some of these frequently asked questions (FAQs).
1. Why are you just focusing on validating a subset of the causal relationships and not all of the arcs predicted by the network inference algorithm?
The goal of the proposed project is to validate a computational method used to predict causal relationships among modeled features from bulk transcriptomic data. Here the features are the prevalence and functional orientation of specific cell types within a tissue and the level of expression of a protein secreted by malignant cells. Causal predictions relate to how a particular feature positively or negatively causes a change in another feature. Validation focuses on testing how well the predicted causal relationships correspond to real-world phenomena under investigation. In short, there are two ways to validate computational predictions, that is using internal or external validation (e.g., [Eddy et al., 2012]). Internal validation is where the overall dataset is divided into two parts, a test set and a validation set. The test set is used to make a prediction and the validation set is used to see if a consistent result is obtained. This is a common approach used in machine learning. This is also how every edge predicted in a graph can be tested using structural intervention distance to assess differences in the structure of causal graphs or cross-entropy to assess faithfulness of the joint probability distribution. The limitation of this approach is that both the test set and the validation data sets are drawn from the same larger data set. This larger data set may not be generalizable to measurements obtained from different experimental systems. Inherent bias in this data set is a common critique of models developed using machine learning (see sexism associated with Amazon’s AI-powered recruitment engine or racism associated with Microsoft’s Tay chatbot).
In addition, subdividing the data set may reduce the statistical power of the test dataset for structural learning. This was one of the observations reported in our previous paper [Kaiser et al., 2016]. To give a bit of context, learning a Bayesian Network involves two parts. First, the structure of the graph is identified, which is comprised of the modeled features or nodes and directed edges or arcs that are statistically enriched in the data set. Second, the identified graph is used to regress the joint probability distribution. The more arcs included in a graph then the more parameters to regress. Overall network inference can be a small n large p problem, where a large number of “p” parameters that describe the relationships among modeled features are constrained by a small number of “n” observations. Of note, the number of parameters grows more than exponentially in relation to the number of features and is related to potential interactions between features, that is an arc between nodes in a network. In engineering, this is called a non-identifiable problem, as you have an insufficient number of observations to theoretically identify the parameters. (Can you uniquely identify the slope and intercept for a linear relationship using one data point? In short, no you can’t.)
Here we use cell deconvolution to reduce the number of features so that there are more observations than features. Cell deconvolution transforms the space of genes expressed in RNA sequencing of a homogenized tissue sample into a cell feature space using patterns of gene expression associated with each cell feature, as a prior. Reducing the number of features more than exponentially reduces the number of parameters. This makes the problem more identifiable, as we have potentially more observations than parameters and, in theory, can identify the parameters. The “in theory” qualifier relates to how informative each observation is. If the data set contains, say, observations that correspond to multiple technical replicates, then the data set is not as informative as a data set that contains observations corresponding to multiple biological replicates. Technical replicates, while they are observations, are not independent samples as are biological replicates. With real data sets, the information content is a bit more difficult to assess. Reducing the number of features associated with a data set will tend to shift a non-identifiable problem towards an identifiable problem. Conversely, increasing the number of features to capture a higher level of granularity in cell-cell communication will make identifiability more difficult. While networks with high granularity may make dense pictures, predictions with lower confidence are likely to lead to more expensive validation experiments that don’t pan out.
The second way to validate a computational prediction is called an external validation, where the model predictions are tested using an entirely different experimental system. This is what we proposed and what we did in the Nature Comm paper [Klinke et al., 2022]. We checked for consistency in the inferred network between bulk transcriptomics data obtained from human breast cancer and human melanoma samples and validated the existence or absence of predicted arcs using syngeneic mouse models for melanoma. We can also compare the predicted arcs against relationships reported in the literature, as discussed in FAQ10 and FAQ11. However, comparing predicted arcs against the literature can suffer from confirmation bias and, as discussed in FAQ4, publication bias. External validation is a more robust validation technique compared to internal validation. However, due to the complexity and experimental tractability of testing specific arcs, not all arcs can be externally validated. We proposed to focus on the arcs related to CCN4, the secreted protein of interest, that has unclear biological mechanisms of action.
2. Are we developing new algorithms to do this network inference?
Developing new algorithms is not a major goal of the project, as there are over 50 deconvolution (e.g., [White et al., 2022]) and 70 network inference [Kitson et al., 2022] algorithms already published. The proposed project focuses on applying existing techniques, namely deconvolution and network inference, to a new problem and combining them in a novel way. It is in this interface that we propose the project. Challenging algorithms in new ways through applying them to new problems can lead to new insights. That said, I (PI Klinke) developed an ensemble network inference approach described in the Nature Comm paper [Klinke et al., 2022], which will be used for this project. Algorithms that discover causal networks from data use different functions to score how well a graph captures the data and different statistical tests for conditional independence between features. The specific scoring functions or statistical tests may introduce systematic inference errors depending on the data considered. Thus an ensemble network inference approach averages out (that is marginalizes) the influence of any single algorithm on the results [Marbach et al., 2010; Marbach et al., 2012].
3. Doesn’t the Bayesian network inference approach have fundamental problems as it can’t find feedback loops, can’t tolerate latent variables, and does not establish causal links?
Instead of toy problems typically used for developing algorithms, focusing on a real application can sharpen the design constraints for the approach proposed. Yes, it is true that there are other algorithms that have the ability to infer feedback loops. However these algorithms require time course data, where pairs of time points are used to “unroll” the feedback loops. The assumption here is that the time course data has been sampled often enough to identify a feedback loop. I discuss sampling frequency in a bit more detail in FAQ6.
Here, though, we focus on tissue samples derived from patients diagnosed with breast cancer. Once a patient is diagnosed with breast cancer, patients are scheduled for surgery to remove the entire primary tumor plus additional tissue depending on the risk of recurrence. Thus the tumor samples are from the patients diagnosed with primary breast cancer and normal samples from uninvolved normal mammary tissue that were obtained following resection. In short, the available data represent single snapshots in time of the tumor microenvironment in each patient. Based on current clinical practice, time course studies are infeasible. However in this project, the central assumption that underpins this approach is that these samples across a larger patient population that share the same disease represent random samples across a common disease trajectory. By generating a dataset where the ground truth is known, we will test this central assumption.
In terms of latent variables, we proposed to test whether there were missing cell subsets in the cell deconvolution approach, as we are using a partial deconvolution approach (we assume cell gene signatures as priors). Ultimately, the directed acyclic graphs (DAGs) are predictions. We propose to validate externally the predictions in Aim 3. This goes along with general maxims about mathematical modeling of systems that bracket the degree of complexity required in a model. These maxims are Occam’s Razor and Einstein’s safety shield. The principle of Occam’s Razor is that you should favor the simplest explanation. The principle of Einstein’s safety shield is that you shouldn’t go simpler. If we are unable to validate externally the model predicted in Aim 1, then the results suggest that we are missing something, potentially a latent variable. This question implies that, a priori, we are missing something. However, without analyzing the data in hand, one can’t make that judgement.
In terms of establishing causal links, the network inference results are predictions (i.e., they are hypotheses about causal links) that require experimental validation. The goal of aim 3 is to validate externally the causal predictions particularly related to the secreted protein, CCN4. As to why we are focused on CCN4, see “Trying not to be a hypocrite” blog post [Klinke, 2022d] and response to FAQ 4.
4. How is this related to cancer immunology?
Identifying collateral targets that can broaden the clinical impact of existing immunotherapies for cancer is a current barrier for progress. Towards that aim, we identified CCN4 in a phenotypic screen for secreted proteins that block an immune signal that is important for organizing an effective anti-tumor immune response. As I discuss in [Old Man Yells at Cloud] blog post [Klinke, 2022a], 90% of papers focus on only 20% of the genome such that the other 80% contain gene products, like the secreted protein CCN4, that are not well characterized [Stoeger et al., 2018]. For instance, it is not clear how CCN4 elicits a cellular response and CCN4 is not included in lists like the RIKEN ligand-receptor connectome [Ramilowski et al., 2015]. Thus, a data-driven approach to identify what cell types are targeted by tumor-secreted CCN4 in humans helps inform a focused experimental strategy to clarify how CCN4 acts. Such information can be used to develop a therapeutic strategy to block its action. We are focused here on CCN4 because we have the experimental systems in place to externally validate the predicted causal arcs. However once validated (which is the point of the proposal), the approach could be applied to other secreted proteins or coding/non-coding RNA that are enriched in extracellular vesicles secreted by malignant cells. As also mentioned in Trying not to be a hypocrite post [Klinke, 2022d], we have also published work related to extracellular vesicles. However, given a clearer path for translation, we have focused here on CCN4.
5. All the cool kids are doing single-cell sequencing/spatial transcriptomics, why don’t you just do that?
Single cell sequencing and spatial transcriptomics are newer experimental techniques and there is a lot of excitement around their use (not to mention a lot of advertising dollars spent). Despite the insight that they provide, there are a number of challenges that remain related to the cost (almost 10 times the cost of bulk sequencing), sparsity (you get fragments of a transcriptomic picture and have to fill in the missing bits), and biases due to batch differences and incomplete tissue digestion. There are solutions to some of these problems, like sparsity and batch correction, that take information from other samples and spread it around. The problem with spreading information around is that potentially biologically important variability gets smoothed out in the process. An important aspect of large scale studies like the Cancer Genome Atlas was standardization, so that samples obtained from different populations obtained at different locations by different researchers can be compared. As echoed in a recent preprint [Hippen et al., 2022], when you start looking at a population instead of a handful of samples, profiling the transcriptome in bulk tumor samples remains the key analytical strategy. Maybe in the future, these issues will be addressed using single-cell methods. The question implies that we should throw out all the information that we have acquired to date about how the tissue microenvironment changes during oncogenesis and wait for some future date when we have accrued an equivalent ensemble of patient samples using these new experimental techniques. Given the clinical need now, our strategy is to combine what information we have now about human populations, that is bulk transcriptomics, plus some targeted experiments to aid in interpreting those existing data.
6. As you propose to use a spontaneous mouse model for breast cancer, isn’t (choose one):
– this mouse model too homogeneous to represent the heterogeneity of human disease?
– the sampling frequency too coarse to fully characterize significant changes in the tumor microenvironment during oncogenesis?
Especially in the area of cancer immunology, mouse models are an important pre-clinical platform to study cause and effect, which is difficult to do in humans. Spontaneous mouse models of cancer are typically genetically primed to develop cancer and are thought to better model oncogenesis. As an alternative, transplantable syngeneic mouse models are commonly used to study how a particular intervention influences tumor progression. Both pre-clinical approaches are designed to be repeatable. To generalize the findings, one would use multiple models, as we have done in our prior work. For instance in [Klinke et al., 2022] we used two cell lines that are used for transplantable models of melanoma, the B16F0 and YUMM1.7 cell lines and here we propose to use spontaneous and transplantable mouse models of breast cancer.
Disease heterogeneity is commonly attributed to genetic variation introduced in the malignant cell. Given the constraint to use mouse models, we selected a spontaneous model as tumors that arise are more genetically diverse than tumors generated using a transplantable model.
While malignant cells may be diverse in humans, we note that the immune system is largely conserved in different individuals. That means that how host immunity is regulated, the cell types that drive the response, and the dynamics of that response can be conserved across individuals. This conservation across individuals underpins vaccination strategies against a defined pathogen, like COVID-19. Yet, the contributions of these different immune components can change during disease progression. Thus disease heterogeneity can also be attributed to variability in disease progression. Some tumors may be detected early while others may be detected late, which can influence the composition of cells within the tumor microenvironment. Here, experiments are designed primarily to observe the heterogeneity associated with disease progression.
An additional challenge with studying humans is that it is difficult to recreate all of these components outside of the individual human. In using in vivo mouse models, all of these components are included and studies can leverage a large body of work describing how mouse immunity is regulated, the cell types that drive the response, and the dynamics. As the question is related to the dynamics, I’d say that the dynamics associated with host immunity is similar between humans and mouse. The proposed sampling scheme is intended to capture timescales from 10 days out to about 1/2 year. In contrast to intracellular signaling that may occur quickly (minutes to hours), the dynamics of changing intercellular networks is a bit slower and more unclear. Though, we know that changes in the prevalence of specific immune cell subsets associated with generating an adaptive immune response takes about 14 days (see [Klinke and Wang, 2017; Wang et al., 2015]). Our sampling scheme will capture that timescale. It is the overall sampling strategy that captures different timescales not a single “optimal” time point. The sampling strategy is related to the Nyquist-Shannon sampling theorem in signal processing.
As an alternative, we also propose to use a Tet-on inducible transplantable mouse model, which we have published on before (see panels J-N of Figure 3 in [Fernandez et al., 2022]). This would allow for knowing when exactly CCN4 was expressed and likely time points that can capture how it changed the cellular network. The drawback of this model is that it is even further afield from the variability of human breast cancer but a pulse-chase experiment is optimal for identifying system dynamics. Or put simply, time-course measurements obtained from a pulse-chase experiment is the traditional approach to establish causal relationships.
7. Network inference (or reverse engineering) is a mature field and a lot of people have been developing network-based models in cancer. So, hasn’t this been done before?
In short, no. As mentioned in the Introduction of our Nature Communication paper [Klinke et al., 2022], the focus of Bayesian network inference to date has been to predict INTRACELLULAR networks, that is how information is passed within a cell to organize a cellular response. In the most cited example, Sachs et al. used a targeted experimental design and flow cytometry to quantify the abundance and phosphorylation state of various intracellular signaling proteins to reconstruct a causal signaling network [Sachs et al., 2005]. As of 2016, Lagani et al. [Lagani et al., 2016] mention that Sachs et al. remains the only case study of Bayesian network learning to reconstruct a causal biological network. One of the reasons being that flow cytometry data provides thousands of independent measures of a couple dozen features, that is n >> p. More recently, single-cell RNAseq data has been used to generate intracellular networks (e.g., [Wang et al., 2021]), which also can be identifiable when the number of samples (n), which here are individual cells, is greater than the number of features, which here are expressed genes (or unique molecular identifiers), considered as part of the intracellular network (p). However, the goal of the project is to validate a computational method used to predict causal networks related to INTERCELLULAR networks, that is how expression of a secreted protein alters the prevalence and functional orientation of different cell types within a tissue.
Current practice for generating causal INTERCELLULAR networks is by hand-curation. While quantitative systems pharmacology (QSP) leaders at Genentech made a similar point regarding hand-curation [Gadkar et al., 2016], a recent 2022 review paper by a QSP working group on the integration of quantitative systems pharmacology and machine learning (ML) [Zhang et al., 2022] states:
QSP modeling is labor-intensive. Model building is still largely performed by manual distillation of a large volume of scientific literature, often by one individual. … the identification of testable mechanistic hypotheses has been widely recognized as one of the most significant challenges due to the general black-box nature of ML approaches [17]. The strength of QSP modeling to address this key weakness of ML, and the strength of data-driven ML to address the QSP weakness of manually building assumptions, suggests that integrated QSP + ML approaches offer the best of both.
In generating a causal directed acyclic graph (DAG), we generate testable mechanistic hypothesis driven by the data, which is illustrated in the Nature Communications article [Klinke et al., 2022]. In terms of structural learning, they go on to give examples of intracellular networks and physics-informed machine learning that implicitly requires time-course data as they are trying to fit a neural network to an ODE model. As we mentioned in FAQ3, data obtained from patient tumor samples are rarely sampled with time. In addition, Certara was tasked by an industry consortium to make a master immune- oncology mechanistic model by integrating together all previously published models, which were collective created by hand-curation [Certara, 2018]. Of note, this aggregate model leaves out cancer associated fibroblasts, which is a highly connected node in our data-driven model.
8. You claim that this is a data-driven approach; yet, you use a blacklist to exclude arcs from being considered as possible arcs in the network. Won’t the blacklist similarly bias the network as hand-curation?
In terms of specification of a blacklist, we discuss this in about three columns of the Nature Communications paper [Klinke et al., 2022]. In short, network inference algorithms consider all possible arcs irrespective of whether they make biological sense. The algorithm may identify two different graphs that equally describe the data in terms of conditional independence, this means that the two graphs exhibit Markov equivalence. One can reject some of the Markov equivalent graphs if they contain arcs named in a blacklist.
In a Bayesian sense, a blacklist can be considered a prior. Instead of a network inference algorithm that is agnostic to current understanding, we can include a small set of specific arcs in a blacklist that don’t make biological sense, like cancer being a leaf node in the resulting cellular network, or that are associated with nodes that have a majority of zero values (i.e., inconsistency with Gaussian assumptions). Cancer being a leaf node in the network implies that a change in the parental feature causes cancer. One example could be an arc implying that a decrease in endothelial cells would cause cancer. This arc does not make biological sense, and so it can safely be blacklisted. Incorporating prior biological knowledge is mentioned in [Lagani et al., 2016] as one of the main factors that enables causal discovery to relate genotype with quantitative traits. So yes, a blacklist does impose a bias but it is not equivalent to hand-curated models. In hand-curated mathematical models, each and every arc is included in the causal network based on a discussion among domain experts. The lack of fibroblasts in the mechanistic immune-oncology model developed by Certara [Certara, 2018] reiterates the point related to the problem with hand-curated models; they only include what experts think are important instead of letting the data speak for themselves. Furthermore, if hand-curated models are the current state-of-the-art in the immuno-oncology space, then we feel that this proposed approach is more data-driven and a significant advance. While we note that there still is some input into the process from domain experts that enters through specifying a minimal set of arcs in a blacklist, we feel that this seems almost negligible compared to a causal network specified entirely by hand. Ultimately, any mechanistic model that is generated must be validated experimentally, which is the focus of Aim 3
9. I see from your biosketch that you, the PI, are an engineer. Why don’t you have a cancer immunologist or immunologist on your team?
While we agree that the project requires a diverse array of skillsets and we have recruited a number of different co-investigators to collaborate on the project, people can evolve their expertise from the degree that they received 25 years ago. As described in a series of blog posts (two “Paper Tales”, [Old Man Yells at Cloud], and “Trying not to be a hypocrite”) [Klinke, 2022b; Klinke, 2022c; Klinke, 2022a; Klinke, 2022d], my professional experiences since I obtained my PhD shape the kind of research questions that we now pursue. That said, my engineering training provided a strong background in computational thinking, rule-based modeling, inverse problem solving – that is trying to infer causal reaction networks from data – and a systems perspective that we are applying to immuno-oncology.
Until July 2022, NIH drew on my scientific expertise as a regular standing member of the Cancer Immunology and Immunopathology study section where I provided expertise in evaluating proposals submitted to NIH related to cancer immunotherapies. In the summer of 2022, the CII and other related study sections were reorganized to balance increased need to review immuno-oncology applications. After this reorganization, I was appointed as a regular member of the Therapeutic Immune Regulation study section, where I now provide expertise in evaluating proposals submitted to NIH related to cancer immunotherapies. Recent publications in 2022 where I am corresponding author, such as the Nature Communications ([Klinke et al., 2022]: first and corresponding author) and EMBO Reports ([Fernandez et al., 2022]: corresponding author) papers, describe experimental results obtained using similar immunocompetent mouse models of cancer as proposed. I am also a member of the American Association of Immunologists, so actually I’m a card-carrying immunologist.
10. The graph predicted from human breast cancer data shows cancer promoting (or a positive causal relationship with) “M1 macrophages” and inhibiting “M2 macrophages”. This seems contradictory to current biological understanding, with “M1 macrophages” being associated with anti-tumor inflammation, and “M2 macro- phages” being associated with immuno-suppression, angiogenesis, neovascularization, and stromal remodeling.
To clarify, cancer is caused by genetic alterations. In many cases, these genetic alterations are caused by environmental factors, like UV radiation in the case of melanoma and mutagens present in cigarette smoke in the case of lung cancers. Each cell has a mechanism to repair DNA upon damage. If the cell is unable to repair the DNA damage, it undergoes programmed cell death that leads to immunosurveillance of malignant clones that also express the same mutated peptides (i.e., immunologic cell death → presentation of mutated antigens and activation of antigen presenting cells, like M1 macrophages → expansion of T cell clones that recognize the mutated antigens). Thus, the default response to acquiring mutations during oncogenesis is an M1 response. By activating and expanding T cells that recognize and kill malignant cells, the M1 response introduces a selective pressure that coupled with continued mutagenesis creates an evolutionary process. Its only after repeated rounds of mutation and selection that tumor cells acquire additional mutations, such as to enhance the expression of CCN4 (see discussion of Table S1 in [Deng et al. 2019]), that enable malignant cells to skew the tissue microenvironment in their favor (immunoediting → immunoescape). Sequencing studies like Pleasance and coworkers in Nature in 2010 [Pleasance et al., 2010a, Pleasance et al., 2010b] and work by Robert Schreiber (e.g., [Dunn et al., 2002]) have elaborated on this process. Thus the prediction that “cancer” promotes (or has positive causal relationship with) M1 macrophages and inhibits M2 macrophages is consistent with current biological understanding of cancer as an evolutionary process.
11. It is unclear how to read out events like the Epithelial-Mesenchymal transition from such causal networks and how they would shift over time. Moreover, it is unclear how cancer directly inhibits mesenchymal cells, but promotes CCN4 that, in turn, promotes mesenchymal cells.
We recently described an unsupervised approach to develop metrics for epithelial versus mesenchymal states of differentiation [Klinke and Torang, 2020]. For epithelial cells, the epithelial state represents full differentiation into an epithelial cell while a mesenchymal state represents a de-differentiation to reacquire a mesenchymal phenotype, which changes morphology and migratory properties. Epithelial-mesenchymal transition (EMT) represents the movement away from an epithelial phenotype and towards a mesenchymal phenotype. Discussions around EMT sometimes imply that cells have a binary switch – that is they are either epithelial or mesenchymal. However, there is an additional view that malignant cells can occupy an intermediate phenotype [George et al., 2017, Jia et al., 2019, Chakraborty et al., 2020], that is a cell doesn’t resemble a fully differentiated epithelial cell but also hasn’t fully reverted to a mesenchymal phenotype. Here, the predicted network suggests that cancer promotes this intermediate phenotype with a slightly greater inhibition of an epithelial state than a mesenchymal state (an effect coefficient of -0.25 for epithelial versus -0.17 for mesenchymal). The network also predicts that the expression of CCN4 drives malignant cells towards a mesenchymal cell state, with an effect coefficient of 1.06. So the malignant transformation of cells (i.e., a pathologist would classify this tissue sample as malignant) is not sufficient to promote full de-differentiation into a mesenchymal cell state but acquiring additional traits, like mutations that increase the expression of CCN4, are required. Of note, the graph depicted in the figure above represents results from both structural learning (the thickness of the arcs depicts the strength of evidence in support of a particular arc) and parameter regression using the inferred DAG (the color of the arcs and linear effect coefficients are annotated on the graph). Normalizing all of the features to the same dynamic range improves interpretability of the effect coefficients.
The local structure associated with the influence of “Cancer” on the “Mesenchymal” state via “CCN4” suggests an incoherent type-3 feed-forward motif to regulate the mesenchymal state. Inference of a feed-forward motif is interesting as feed-forward loops are highly prevalent and well understood as control mechanisms in intracellular networks [Alon, 2007] but are less well understood in the context of intercellular networks. This was discussed in our Nature Communications article. Feed-forward motifs are interesting as they can temporally disconnect a biological dose from a response. This is important as the top use of QSP models in immuno-oncology is for dose-scheduling [Lemaire et al., 2022]. This network prediction then motivates targeted validation experiments. Disconnecting dose-response relations can have important implications for drug discovery and development.
References
- [Alon, 2007] Alon, U. (2007). Network motifs: theory and experimental approaches. Nat Rev Genet, 8(6):450–461.
- [Certara, 2018] Certara (2018). Certara launches industry-first quantitative systems pharmacology (QSP) consortium on immuno-oncology with leading pharma company members. https://preview.tinyurl.com/yaxb4hmw. Accessed: 2020-05-18.
- [Chakraborty et al., 2020] Chakraborty, P., George, J. T., Tripathi, S., Levine, H., and Jolly, M. K. (2020). Comparative Study of Transcriptomics-Based Scoring Metrics for the Epithelial-Hybrid-Mesenchymal Spectrum. Front Bioeng Biotechnol, 8:220.
- [Dastin, 2018] Dastin, J. (2018). Amazon scraps secret ai recruiting tool that showed bias against women. https://www.reuters.com/article/us-amazon-com-jobs-automation-insight/amazon-scraps-secret-ai-recruiting-tool-that-showed-bias-against-women-idUSKCN1MK08G. Accessed: 2023-01-09.
- [Deng et al., 2019] Deng, W., Fernandez, A., McLaughlin, S. L., and Klinke, D. J. (2019). WNT1-inducible signaling pathway protein 1 (WISP1/CCN4) stimulates melanoma invasion and metastasis by promoting the epithelial-mesenchymal transition. J. Biol. Chem., 294(14):5261–5280.
- [Dunn et al., 2002] Dunn, G. P., Bruce, A. T., Ikeda, H., Old, L. J., and Schreiber, R. D. (2002). Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol, 3(11):991–998.
- [Eddy et al., 2012] Eddy, D. M., Hollingworth, W., Caro, J. J., Tsevat, J., McDonald, K. M., and Wong, J. B. (2012). Model transparency and validation: a report of the ISPOR-SMDM Modeling Good Research Practices Task Force-7. Med Decis Making, 32(5):733–743.
- [Fernandez et al., 2022] Fernandez, A., Deng, W., McLaughlin, S. L., Pirkey, A. C., Rellick, S. L., Razazan, A., and Klinke, D. J. (2022). Cell Communication Network factor 4 promotes tumor-induced immunosuppression in melanoma. EMBO Rep, page e54127.
- [Gadkar et al., 2016] Gadkar, K., Kirouac, D. C., Mager, D. E., van der Graaf, P. H., and Ramanu- jan, S. (2016). A Six-Stage Workflow for Robust Application of Systems Pharmacology. CPT Pharmacometrics Syst Pharmacol, 5(5):235–249.
- [George et al., 2017] George, J. T., Jolly, M. K., Xu, S., Somarelli, J. A., and Levine, H. (2017). Survival Outcomes in Cancer Patients Predicted by a Partial EMT Gene Expression Scoring Metric. Cancer Res., 77(22):6415–6428.
- [Hippen et al., 2022] Hippen, A. A., Omran, D. K., Weber, L. M., Jung, E., Drapkin, R., Doherty, J. A., Hicks, S. C., and Greene, C. S. (2022). Performance of computational algorithms to deconvolve heterogeneous bulk tumor tissue depends on experimental factors. bioRxiv.
- [Jia et al., 2019] Jia, D., George, J. T., Tripathi, S. C., Kundnani, D. L., Lu, M., Hanash, S. M., Onuchic, J. N., Jolly, M. K., and Levine, H. (2019). Testing the gene expression classification of the EMT spectrum. Phys Biol, 16(2):025002.
- [Kaiser et al., 2016] Kaiser, J. L., Bland, C. L., and Klinke, D. J. (2016). Identifying causal networks linking cancer processes and anti-tumor immunity using Bayesian network inference and metagene constructs. Biotechnol. Prog., 32(2):470–479.
- [Kitson et al., 2022] Kitson, N. K., Constantinou, A. C., Guo, Z., Liu, Y., and Chobtham, K. (2022). A survey of bayesian network structure learning. arXiv
- [Klinke, 2022a] Klinke, D. J. (2022a). [old man yells at cloud]. https://dkdesignwv.com/2022/09/09/old-man-yells-at-cloud/. Accessed: 2023-01-09.
- [Klinke, 2022b] Klinke, D. J. (2022b). Paper tales Klinke DJ Journal of Theoretical Biology 2006. https://dkdesignwv.com/2022/08/20/paper-tales-klinke-dj-journal-of-theoretical-biology-2006/ Accessed: 2023-01-09.
- [Klinke, 2022c] Klinke, D. J. (2022c). Paper tales Klinke DJ PLoS One 2008. https://dkdesignwv.com/2022/08/26/paper-tales-klinke-dj-plos-one-2008/ Accessed: 2023-01-09.
- [Klinke, 2022d] Klinke, D. J. (2022d). Trying to not be a hypocrite. https://dkdesignwv.com/2022/09/16/trying-to-not-be-a-hypocrite-warning-science-language/ Accessed: 2023-01-09.
- [Klinke et al., 2022] Klinke, D. J., Fernandez, A., Deng, W., Razazan, A., Latifizadeh, H., and Pirkey, A. C. (2022). Data-driven learning how oncogenic gene expression locally alters heterocellular networks. Nat Commun, 13(1):1986.
- [Klinke and Torang, 2020] Klinke, D. J. and Torang, A. (2020). An Unsupervised Strategy for Identifying Epithelial-Mesenchymal Transition State Metrics in Breast Cancer and Melanoma. iScience, 23(5):101080.
- [Klinke and Wang, 2017] Klinke, D. J. and Wang, Q. (2017). Inferring the Impact of Regulatory Mechanisms that Underpin CD8+ T Cell Control of B16 Tumor Growth In vivo Using Mechanistic Models and Simulation. Front Pharmacol, 7:515.
- [Lagani et al., 2016] Lagani, V., Triantafillou, S., Ball, G., Tegn ́er, J., and Tsamardinos, I. (2016). Probabilistic Computational Causal Discovery for Systems Biology, in Uncertainty in Biology: A Computational Modeling Approach, pages 33–73. Springer.
- [Lemaire et al., 2022] Lemaire, V., Bassen, D., Reed, M., Song, R., Khalili, S., Lien, Y. T. K., Huang, L., Singh, A. P., Stamatelos, S., Bottino, D., and Hua, F. (2022). From Cold to Hot: Changing Perceptions and Future Opportunities for Quantitative Systems Pharmacology Modeling in Cancer Immunotherapy. Clin Pharmacol Ther.
- [Marbach et al., 2012] Marbach, D., Costello, J. C., Ku ̈ffner, R., Vega, N. M., Prill, R. J., Camacho, D. M., Allison, K. R., Kellis, M., Collins, J. J., Stolovitzky, G., Aderhold, A., Allison, K. R., Bonneau, R., Camacho, D. M., Chen, Y., Collins, J. J., Cordero, F., Costello, J. C., Crane, M., Dondelinger, F., Drton, M., Esposito, R., Foygel, R., de la Fuente, A., Gertheiss, J., Geurts, P., Greenfield, A., Grzegorczyk, M., Haury, A. C., Holmes, B., Hothorn, T., Husmeier, D., Huynh- Thu, V. A., Irrthum, A., Kellis, M., Karlebach, G., Ku ̈ffner, R., L`ebre, S., De Leo, V., Madar, A., Mani, S., Marbach, D., Mordelet, F., Ostrer, H., Ouyang, Z., Pandya, R., Petri, T., Pinna, A., Poultney, C. S., Prill, R. J., Rezny, S., Ruskin, H. J., Saeys, Y., Shamir, R., Sˆırbu, A., Song, M., Soranzo, N., Statnikov, A., Stolovitzky, G., Vega, N., Vera-Licona, P., Vert, J. P., Visconti, A., Wang, H., Wehenkel, L., Windhager, L., Zhang, Y., and Zimmer, R. (2012). Wisdom of crowds for robust gene network inference. Nat. Methods, 9(8):796–804.
- [Marbach et al., 2010] Marbach, D., Prill, R. J., Schaffter, T., Mattiussi, C., Floreano, D., and Stolovitzky, G. (2010). Revealing strengths and weaknesses of methods for gene network inference. Proc. Natl. Acad. Sci. U.S.A., 107(14):6286–6291.
- [Mason, 2016] Mason, P. (2016). The racist hijacking of microsoft’s chatbot shows how the internet teems with hate. https://www.theguardian.com/world/2016/mar/29/microsoft-tay-tweets-antisemitic-racism Accessed: 2023-01-09.
- [Pleasance et al., 2010a] Pleasance, E. D., Cheetham, R. K., Stephens, P. J., McBride, D. J., Humphray, S. J., Greenman, C. D., Varela, I., Lin, M. L., ez, G. R., Bignell, G. R., Ye, K., Alipaz, J., Bauer, M. J., Beare, D., Butler, A., Carter, R. J., Chen, L., Cox, A. J., Edkins, S., Kokko-Gonzales, P. I., Gormley, N. A., Grocock, R. J., Haudenschild, C. D., Hims, M. M., James, T., Jia, M., Kingsbury, Z., Leroy, C., Marshall, J., Menzies, A., Mudie, L. J., Ning, Z., Royce, T., Schulz-Trieglaff, O. B., Spiridou, A., Stebbings, L. A., Szajkowski, L., Teague, J., Williamson, D., Chin, L., Ross, M. T., Campbell, P. J., Bentley, D. R., Futreal, P. A., and Stratton, M. R. (2010a). A comprehensive catalogue of somatic mutations from a human cancer genome. Nature, 463(7278):191–196.
- [Pleasance et al., 2010b] Pleasance, E. D., Stephens, P. J., O’Meara, S., McBride, D. J., Meynert, A., Jones, D., Lin, M. L., Beare, D., Lau, K. W., Greenman, C., Varela, I., Nik-Zainal, S., Davies, H. R., ez, G. R., Mudie, L. J., Latimer, C., Edkins, S., Stebbings, L., Chen, L., Jia, M., Leroy, C., Marshall, J., Menzies, A., Butler, A., Teague, J. W., Mangion, J., Sun, Y. A., McLaughlin, S. F., Peckham, H. E., Tsung, E. F., Costa, G. L., Lee, C. C., Minna, J. D., Gazdar, A., Birney, E., Rhodes, M. D., McKernan, K. J., Stratton, M. R., Futreal, P. A., and Campbell, P. J. (2010b). A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature, 463(7278):184–190.
- [Ramilowski et al., 2015] Ramilowski, J. A., Goldberg, T., Harshbarger, J., Kloppmann, E., Klopp- man, E., Lizio, M., Satagopam, V. P., Itoh, M., Kawaji, H., Carninci, P., Rost, B., and Forrest, A. R. (2015). A draft network of ligand-receptor-mediated multicellular signalling in human. Nat Commun, 6:7866.
- [Sachs et al., 2005] Sachs, K., Perez, O., Pe’er, D., Lauffenburger, D. A., and Nolan, G. P. (2005). Causal protein-signaling networks derived from multiparameter single-cell data. Science, 308(5721):523–529.
- [Stoeger et al., 2018] Stoeger, T., Gerlach, M., Morimoto, R. I., and Nunes Amaral, L. A. (2018). Large-scale investigation of the reasons why potentially important genes are ignored. PLoS Biol, 16(9):e2006643.
- [Wang et al., 2015] Wang, Q., Klinke, D. J., and Wang, Z. (2015). CD8(+) T cell response to adenovirus vaccination and subsequent suppression of tumor growth: modeling, simulation and analysis. BMC Syst Biol, 9:27.
- [Wang et al., 2021] Wang, X., Choi, D., and Roeder, K. (2021). Constructing local cell-specific networks from single-cell data. Proc Natl Acad Sci U S A, 118(51).
- [White et al., 2022] White, B. S., de Reyni`es, A., Newman, A. M., Waterfall, J. J., Lamb, A., Petitprez, F., Valdeolivas, A., Lin, Y., Li, H., Xiao, X., Wang, S., Zheng, F., Yang, W., Yu, R., Guerrero-Gimenez, M. E., Catania, C. A., Lang, B. J., Domanskyi, S., Bertus, T. J., Pier- marocchi, C., Monaco, G., Caruso, F. P., Ceccarelli, M., Yu, T., Guo, X., Coller, J., Maecker, H., Duault, C., Shokoohi, V., Patel, S., Liliental, J. E., Simon, S., Saez-Rodriguez, J., Heiser, L. M., Guinney, J., and Gentles, A. J. (2022). Community assessment of methods to deconvolve cellular composition from bulk gene expression. bioRxiv.
- [Zhang et al., 2022] Zhang, T., Androulakis, I. P., Bonate, P., Cheng, L., Helikar, T., Parikh, J., Rackauckas, C., Subramanian, K., Cho, C. R., Androulakis, I. P., Bonate, P., Borisov, I., Broderick, G., Cheng, L., Damian, V., Dariolli, R., Demin, O., Ellinwood, N., Fey, D., Gulati, A., Helikar, T., Jordie, E., Musante, C., Parikh, J., Rackauckas, C., Saez-Rodriguez, J., Sobie, E., Subramanian, K., and Cho, C. R. (2022). Two heads are better than one: current landscape of integrating QSP and machine learning : An ISoP QSP SIG white paper by the working group on the integration of quantitative systems pharmacology and machine learning. J Pharmacokinet Pharmacodyn, 49(1):5–18.
