

Creating a mathematical model of disease based on the available literature requires reading a lot of papers. In the 5 years that I was at Entelos, I had read close to 3000 papers. In the early 2000’s, these were largely still on paper and involved someone going to the stacks at Stanford University library and copying what we wanted to read. By “reading”, I mean digging into the paper looking for data that we could use to calibrate/validate the model against. Many times these data were not mentioned in the abstract or key findings of the paper but appeared in control or time-course experiments. From my current perspective of West Virginia, we mined the scientific seams of literature for data.
In the 20 years since, some things have changed, others remain the same. While access to scientific papers and the sharing of raw data has vastly improved, the practice of science remains largely the same. The bread-and-butter publication in basic science is the product of a PhD student. In the process of defining their dissertation, the PhD student narrows the scope of their proposed project to focus on the key question and to graduate in a timely fashion. Experiments are then simplified to have a single experimental variable. Measurements are acquired at a single time point and are analyzed using null hypothesis tests. If the results are deemed to be statistically significant, their analysis suggests that the results observed after the designed intervention is not explained by random chance. Additional experiments may be performed to help explain the key finding, typically to provide a mechanistic explanation for a statistically significant result. While these bread-and-butter publications provided the data foundation for the mechanistic mathematical models of disease that we developed at Entelos, they also presented challenges for building these models particularly related to signal integration, heterocellular signaling within tissues, and observational bias.
Signal integration
In short, atopic asthma is an immune-mediated disease of the lung whereby inhalation of some seemingly innocuous substance, like mold or cat hair, causes a biphasic reduction in lung function – an immediate reduction starting within minutes followed by an improvement and subsequent reduction 8-12 hours later. In creating the model, we modeled key cell types that are involved in the disease and the secreted factors that they release. Cell types typically release a variety of secreted factors and each secreted factor has an influence on multiple cell types. For instance, mast cells release pre-formed granules when triggered with an appropriate antigen. These granules enable the rapid release of histamine and two enzymes: chymase and tryptase. Histamine, in turn, influences the contractile state of smooth muscle cells that can reduce the circumference of the airway, releases mucus into the airways by goblet cells that line the airways, and influences the vascular permeability of endothelial cells causing swelling of the airway tissue. The biochemical trigger that initiates the release of granules also initiates the synthesis and release of lipid mediators like cysteinyl leukotrienes and PGD2. Cysteinyl leukotrienes also influence vascular permeability; Cysteinyl leukotriene and PGD2 influence smooth muscle contraction; and Cysteinyl leukotrienes, PGD2 and chymase contribute to mucus secretion. The overlapping functions of these biochemical mediators, yet differing secretion dynamics, implies that their relative importance in the early versus late phase of the lung function response is different. A drug that targets only one of these biochemical mediators, like an anti-histamine, may provide relief to the early phase but not the late phase.
A snapshot of the Asthma PhysioLab showing that the biochemical mediators PAF, fibronectin, TNF-alpha, IL3, GMCSF, and IL5 promote while TGF-beta inhibits the activation of tissue eosinophils.
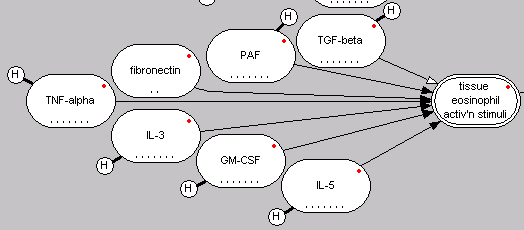
Basic science papers, though, typically focus on characterizing isolated parts of this system (see [1] as similar critique), such as the action of one biochemical mediator on smooth muscle contraction. The challenge in integrating all data describing the behavior of the isolated parts is determining how a cell integrates these different signals. Does a cell add them, is there a total maximum, are they added equally, or do some mediators potentiate the response of others? These are all questions that come to the forefront when trying to build a mechanistic mathematical model that integrates these data. Additional constraints, like clinical studies of lung function measurements reported in time and in response to existing therapies, help narrow down some of the options. However uncertainty remains as it is a characteristic weakness of how we study integrated systems, like a tissue.
Party in the house tissue
Like a good party, normal function of a tissue is maintained in the presence of external challenges through the efficient flow of information between different cell types present within that tissue, like the lung. In contrast to man-made systems, biological systems present challenges for identifying how components of the system causally influence each other. In engineering, causal relationships between system components are inferred from a set of input cues and output responses. In context of atopic asthma, an input cue may be a drug that neutralizes histamine and an output response may be lung function. Many approaches exist for identifying simple-input-simple-output (SISO) systems – where a change in input causes a unique change in output. However, approaches for identifying causal relationships among components of more complex integrated closed-loop systems, like a tissue, are less well developed.
A closed-loop system is defined as a multi-component system where the output of one component provides the input to another component. Closed-loop systems, like tissues, are particularly challenging as it is impossible to identify the relationships among cells of a system based upon overall input (e.g. drugs) and output (e.g. tissue function) measurements. One of the reasons for this is that changes in the internal state of the system may alter the response of the system to a defined input, such that there is not a direct causal relationship between overall system input and output. Historically, the causal mechanisms underlying the behavior of closed-loop systems in physiology have been identified via ingenious methods for isolating elements within the integrated system (i.e., “opening the loop”).
A classic example of opening the loop where the whole body is the closed-loop system is the discovery of insulin and its’ role in connecting food intake to substrate metabolism. As insulin is only produced by the endocrine pancreas, measuring plasma insulin provides a direct measure of the communication between organs that relate food intake (occurs in the intestines) and substrate metabolism in the peripheral tissues (like adipose tissue and muscle). We can then reduce the integrated system into a set of connected subsystems, like the endocrine pancreas. The pancreas can then be approximated as a SISO subsystem where the glucose concentration in the portal vein is the input and insulin release into the plasma is the output. Measuring changes in insulin in the blood in response to changes in plasma glucose provide the basis for partitioning alterations in system response (e.g., diabetes) into deficiencies in insulin production (i.e., type 1 diabetes) and insulin action (i.e., type 2 diabetes). Treatment for diabetes is tailored to the deficiency in component function that exists in the patient. In diabetes, “opening the loop” means identifying organ-level cross-talk using blood measurements. In contrast, the cell-level cross-talk between different cell types within tissues requires a more sophisticated experimental design than just measuring the number of cell types present. There are some single-cell methods – like CellPhoneDB [2] – that are trying to address this, but this leads to the third point.
Observational bias
In clarifying our understanding of the biology that we wanted to model, we typically consulted with leading experts in the field. We would read the literature and prepare questions for consulting visits. A common theme that emerged from these conversations is that there are significant gaps in our understanding of biology. As one put it: we study what we know we can measure (a.k.a., the streetlight effect ). As each discipline has their own tried-and-true methods to measure aspects of biological systems, where does that collectively leave us? Well, a recent survey of the literature reported that over 90% of scientific publications focus on only 20% of the genome [3]. I would assume that 80% of the genome is there for a reason, but we just don’t clearly know why.
In addition to not knowing what 80% of the genome does, we also assume that cell-to-cell communication occurs through the interaction of two molecular entities, such as a protein secreted by one cell type (e.g., insulin) interacting with a transmembrane protein expressed by another cell type (e.g., the insulin receptor) or a transmembrane protein on one cell interacting with a transmembrane protein on another cell. We have lots of methods to measure individual proteins. But less-biased ways of measuring biomolecules have revealed additional modes of communication, such as through the cellular release of extracellular vesicles. Extracellular vesicles are nano-scaled structures that contain proteins and coding and non-coding RNA. If single proteins used for cell-to-cell communication are considered as individual words, these extracellular vesicles could promote more complicated communication in the form of whole paragraphs. The roles that these extracellular vesicles play in mediating cell-to-cell communication within tissues is just tentatively emerging.
If understanding 20% of the genome is sufficient to develop effective drugs to treat disease, then we’re good. However we’ve been fighting the “War on Cancer” since the 1970s and there many patients that are underserved by the current treatment options. To mix metaphors: continually looking under the streetlight and expecting a cure for cancer, isn’t that the definition of insanity? Of note cancer is a disease caused by genetic alterations. As every cell can potentially re-express every gene in the genome, these genetic alterations can re-introduce a gene product out of it’s evolutionarily designed context. Thus, our biological knowledge constructed from the literature, which in aggregate represents normal biology, may not apply.
Then if everyone is looking at 20% of the genome, one strategy to make progress is to create a system where you have the potential to look outside of that area. In numerical integration, antithetic sampling is a similar idea used to accelerate convergence. But you don’t need a PhD to know that; if you like to find eggs at an Easter Egg hunt, you don’t follow the crowd. In a future post, I’ll talk about our approach.
Gestalt
To me, the most valuable part of building a mechanistic mathematical model is the process. To the company, it was the product. The process of formulating a mechanistic model of pathophysiology really helps clarify what we do and don’t understand about a particular disease area. In short, it forces you to connect the dots in a quantitative way. This is really what separates mechanistic modeling from, say, bioinformatics, which both rely on mathematics and a computer and involve biological data.
The focus on the modeling product is a bit problematic. When you make a model, whether it be a mathematical or a biological one, you are abstracting the real system to create an artificial system that only contains what you think are the key elements of the real system. Everything else is removed. We then use these models to ask questions that we can’t or are difficult to ask in the real system. In order to extrapolate the results obtained from the model system to inform our understanding of the real system, we need to know whether the question that we asked of the model impinges on the elements that we thought to be not important. The more simplified the model (like a cancer cell line in a dish), the more you leave out (e.g., immune system, blood flow, stromal cells, different cancer clones). Maybe that’s ok, but you need to really understand the biological context that you’re trying to model. When you try to sell a mechanistic mathematical model as a shrink-wrapped product, you are missing the context. In my view, that is why Entelos’ original business model failed. To do this work in academia can be difficult, as the enabling skills are associated with different disciplines. More on that later.
References
[1] Lazebnik, Y. Can a biologist fix a radio?–Or, what I learned while studying apoptosis. Cancer Cell (2002) 2, 179–182.
[2] Efremova M, Vento-Tormo M, Teichmann SA, Vento-Tormo R. CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nature Protocols (2020) 15:1484–1506.
[3] Stoeger T, Gerlach M, Morimoto RI, Nunes Amaral LA. Large-scale investigation of the reasons why potentially important genes are ignored. PLoS Biol (2018) 16(9): e2006643.
